Blog
Identifying Causal Variants in Mendelian Diseases: Common Pitfalls and Strategies
Key Takeaways
- In Mendelian disorders, identifying the causal variant is crucial for accurate research into genetic diseases
- Incorrect variant annotation, incomplete penetrance, and phenocopies are common pitfalls in the identification of causal variants
- Careful interpretation of genetic data and consideration of multiple lines of evidence are necessary to avoid these pitfalls and improve research accuracy
- Strategies for improving research accuracy and success rates include using multiple genetic testing methods which overlap the full spectrum of variation such as sequencing and optical genome mapping

The recent publication by Al Abdi et al. 2023 in Nature Communications is the largest study of its kind on Mendelian disorders. In this publication, the authors analyzed data from 4,577 families with genetic diseases and identified several common pitfalls in the identification of causal variants, including incorrect variant annotation, incomplete penetrance, and phenocopies. This publication is of particular importance as it highlights the challenges in identifying causal variants in genetic diseases and the potential consequences of misinterpretation which affect geneticists and patients worldwide and can lead to long diagnostic odysseys. They also discuss the implications of these pitfalls for genetic diagnosis and counseling. This article is important because it highlights the need for careful interpretation of genetic data and the importance of considering multiple lines of evidence as well as genetic analysis methods such as optical genome mapping (OGM) when conducting genetic disorder research.
Genetic diseases are caused by mutations in genes that can be inherited or arise spontaneously, with Mendelian diseases being caused by mutations in a single gene. Identifying the causal variant is crucial for accurate diagnosis and treatment of genetic disease. However, despite the remarkable advances in both disease genetics and technologies such as sequencing, across all indications exome sequencing (WES) cohorts typical report a <50% diagnostic rate,1 whereas in suspected Mendelian diseases a recent real-world study on the clinical implementation of Whole Genome Sequencing (WGS) reported a 35% diagnostic rate falling to 11% for complex cases.2 Therefore, it is clear that despite the technologies and analysis tools available, there are persistent challenges in the understanding of genetic disorders, and specifically Mendelian disorders, which traverse individual samples and uniformly affect large genomic programs. In this publication the study authors examine these challenges within a large Mendelian genomic programme in the Kingdom of Saudi Arabia.
Identified issues with the cohort
Within this publication, several challenges for identifying causal variants were identified which can be broadly broken into four areas as shown in Figure 1. The authors estimated a probability of 34.3% for encountering at least one of these challenges.
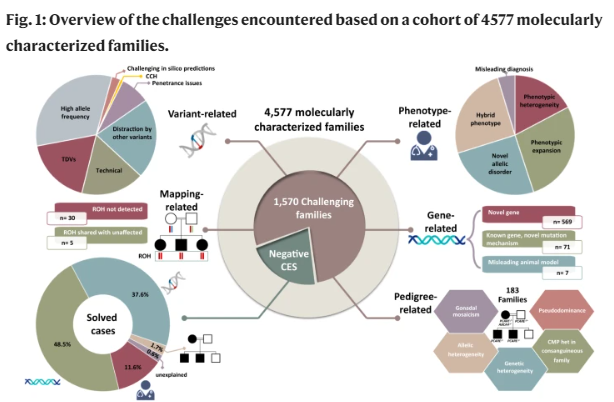
- Phenotype related
- Phenotype Heterogeneity (3% of Families) – The phenotype was sufficiently heterogeneous (intrafamilial or interfamilial) to complicate the original molecular diagnosis
- Phenotype Expansion (5% of families) – The phenotype provided by the referring physician was sufficiently different from the typical phenotypic expression of the implicated gene to make the molecular diagnosis challenging
- Allelism (5.3% of families) – The phenotype is sufficiently different from the one described in the literature that it justifies labeling as a distinct allelic disorder
- Blended Phenotype (5.5% of families) – Phenotypes caused by the presence of two or more Mendelian diseases in the same individual
- Erroneous clinical labels (1% of families) – The wrong diagnostic label caused a major delay in the molecular diagnosis
- Non-Mendelian phenotypes (1.8% of families) – These include neurodevelopmental disorders that turned out to be caused by environmental factors unknown at the time of recruitment and familial clustering of complex phenotypes that masquerade as Mendelian phenocopies
- Gene Related
- Novel gene-disease assertions (9.8% of families) – When the gene-disease assertion was novel at the time of analysis, the molecular diagnosis was greatly delayed
- Incompatible phenotype in the animal model (0.45% of families) – Where the reported phenotype in the animal model is sufficiently incompatible to delay the establishment of the gene-disease assertion as the phenotype of the animal model is an important consideration when assessing novel gene-disease associations
- Known gene, novel mutation mechanism (4.5% of families) – The likely causal variant was dismissed because of the perceived incompatibility of the identified homozygous variant with the established dominant inheritance pattern of the implicated gene
- Variant Related
- Interpretation challenges
- Tentative transcript-deleterious variants (TDV) and Allele frequencies above cut off
- The variant is a founder variant that causes a disease with a previously unrecognized relatively high incidence
- The variant is only pathogenic when compound heterozygous with a more damaging variant
- The variant leads to a highly variable phenotype
- The variant causes a common but embryonic lethal disease
- The variant causes a common but cryptic phenotype
- No clear reason for the discrepancy between the high AF and the observed disease phenotype – this can be due to challenging in silico prediction, complex compound inheritance, multivariant alleles, incomplete penetrance, red herring variants and presumptive loss of function in known disease genes
- Loss of function is not a disease mechanism
- The variant is not a true loss of function
- The reported gene-disease assertion is refuted
- Technical challenges
- Deep intronic variants – Not typically covered by WES
- Regulatory elements
- Repeat expansions – Not well covered by sequencing
- Larger genomic rearrangements – (Variants >50 bp in size) but below the limit of detection of chromosomal microarray were challenging to call on exome sequencing but these were resolved with OGM
- Pseudogenes
- Platform and bioinformatic limitations – Missing or miscalled due to platform
- Epigenetic – Not covered by sequencing
- Pedigree Related
- Pseudodominance
- Gonadal mosaicism
- Allelic and genetic heterogeneity
Impact of these challenges
This publication highlights several common pitfalls in the identification of causal variants in genetic diseases. Incorrect variant annotation can lead to misinterpretation of the data and incorrect identification of pathogenic variants. Incomplete penetrance occurs when not all individuals with the mutation exhibit the disease phenotype, which can complicate understanding of the genetic conditions. Phenocopies are non-genetic conditions that mimic the symptoms of a genetic disease, leading to incorrect identification of pathogenic causes of a genetic condition. These pitfalls can be avoided by careful interpretation of genetic data and consideration of multiple lines of evidence.
The study authors reanalyzed and reinterpreted 314 samples with negative clinical exome or genome sequencing using a combination of traditional and novel techniques and were able to identify the likely causal variant in 54.5% of samples. OGM was used to analyze samples that remained negative after ES and was particularly relevant in the detection of large variants that were below the limit of detection of chromosomal microarray (CMA).
The authors of this study concluded that their findings highlight the need for a thorough approach to cases that remain unresolved after sequencing. Strategies for improving accuracy include using multiple genetic testing methods, such as WES, WGS and non-sequencing techniques such as chromosomal microarray and OGM, as well as specifically considering non-genetic factors such as family history and clinical presentation.
OGM’s utility
In this study, OGM was utilized on selected negative cases following WES to leverage its ability to capture structural variant (SV) and copy number information that could not be obtained by sequencing or CMA alone. The effectiveness of OGM was demonstrated in a case involving a pedigree structure, where researchers examined the difficulties tied to imprinting disorders that seemed to present autosomal recessive traits. OGM was employed to investigate three pediatric instances of split hand/foot malformation syndrome when karyotyping, ES, and RNA sequencing were unable to identify a causal variant. OGM detected a tandem duplication affecting several genes in all three cases, suggesting paternal gonadal mosaicism. Beyond this discovery, the authors noted additional cases where OGM was used to solve highly complex cases involving SVs from various classes.
This research, the most comprehensive of its kind on Mendelian disorders, emphasizes that the definitive molecular diagnosis of diseases is fraught with complexities stemming from the data generation process itself. The authors illustrate that employing a mix of techniques can uncover more information, potentially enhancing the understanding of Mendelian diseases. They also highlight the potential of OGM to tackle intricate cases involving SVs, suggesting that OGM could be instrumental in addressing the variant detection gap left by other methods. The results of this multifaceted approach, which incorporates several orthogonal methods, provide researchers with a rich data set that could lead to an increase in the understanding of Mendelian diseases.
For more information on how OGM can be applied to genetic disease research, please visit our webpage.




