Blog
Streamline Your Analysis for Swift Results with VIA™ Software

VIA revolutionizes data annotation, filtering, and interpretation by automating these processes, drastically reducing the time spent manually navigating databases. Accelerating the path to results, VIA software ensures rapid and improved classification of variants by pathogenicity while minimizing associated time and costs.
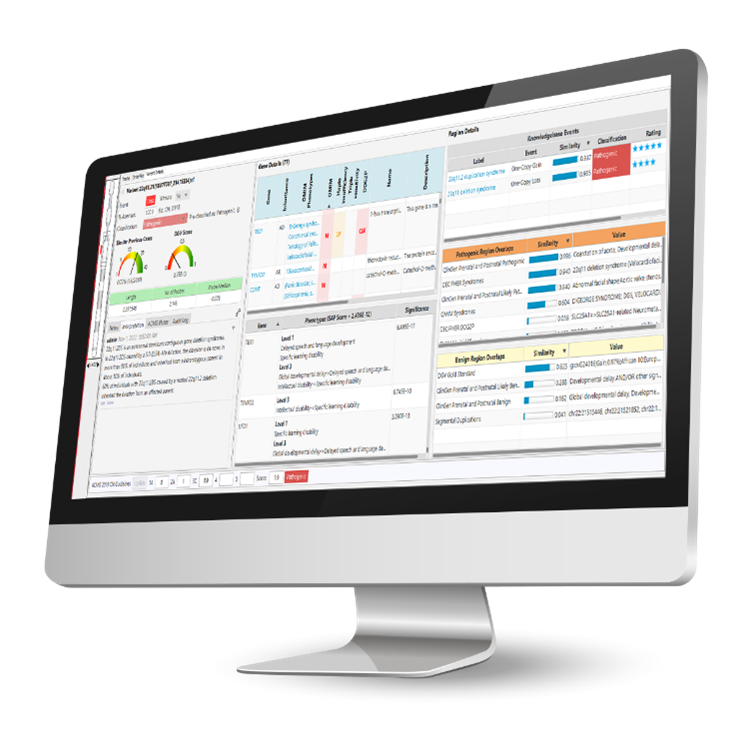
Effortlessly make informed variant interpretations with VIA’s intelligent automation for variant classification that utilizes consolidated gene/variant annotations from both internal and external databases. VIA employs a rules-based decision tree for event pre-classification, and its dynamic variant filters, encompassing panels, event-level details, and frequency information, facilitate the swift identification of pivotal variants. Moreover, VIA software provides access to historical cases, enabling the retrieval of information from prior samples and the establishment of a local knowledge base housing expert curations for future reference.
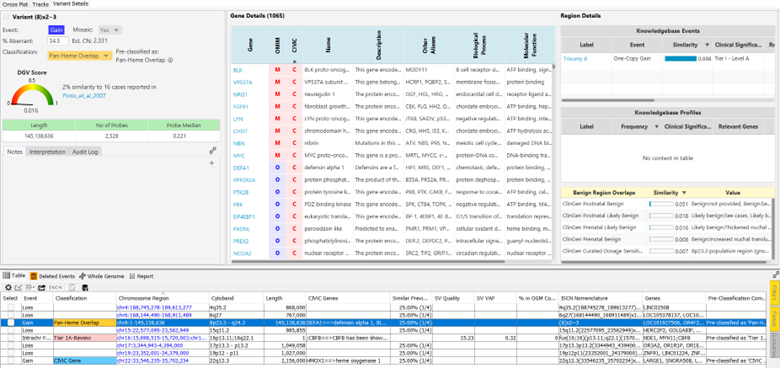
Experience VIA’s capabilities to enrich your analysis workflow across application areas to tailor the automation assisted interpretations to the analysis objectives, guiding analysts toward likely pathogenic findings. Enhance constitutional case analysis further by incorporating sample phenotypes. VIA software leverages phenotypic information to construct a sample-specific gene panel based on comprehensive gene-phenotype associations, using Human Phenotype Ontology (HPO) terms. This approach offers a prioritization metric, facilitating a focused examination of relevant events to quickly bring the most important variants to the forefront. VIA software supports the interpretation process by presenting a shortlist of variants with detailed information from both internal and external resources. Gain insights into frequency, gene details, and Bionano curated annotation resources, all in a single view. Each copy number variant (CNV) is evaluated using an integrated calculator aligned with ACMG standards for CNV pathogenicity scoring, resulting in a numeric assertion of pathogenicity saved with the case data for completeness and future reference.
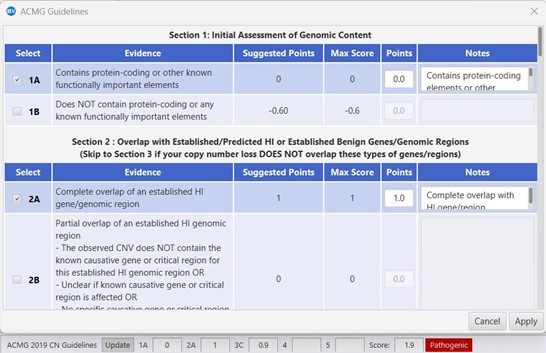
Leverage VIA’s capabilities to avoid duplicating high-value knowledge collection and curation efforts. Database calls include classification status, aiding in the quick identification of similar events detected previously. Record curated interpretations for variants and genes in an internal knowledge base for seamless access to local curated knowledge, expediting the case analysis process and ensuring consistency with written interpretations.
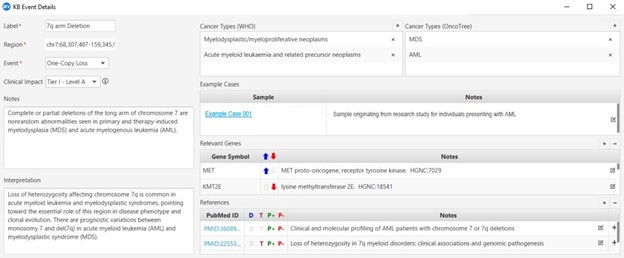
VIA’s functionality extends to efficiently ruling out benign events, as well. Analysts can tap into supportive information on benign impact based on observed frequency within internal and external databases that inform automated classifications. Automatically cataloging benign variants is important for constructing a valuable resource to understand common human variation and ensuring comprehensive analyses.
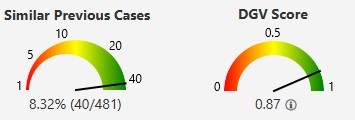
In conclusion, VIA software offers robust support for variant interpretation, enabling a swift focus on relevant variants and a comprehensive understanding of their impact on disease. Elevate your cytogenomic variant interpretation with VIA software for faster and more accurate case analyses.



