Blog
Shallow Sequencing (Low-Pass) CNV Analysis: A Brief Guide
Copy number variations (CNVs) are genomic alterations that result in abnormal copies of one or more genes. Structural genomic events such as duplications, deletions, translocations, and inversions can cause CNVs.
Similar to single-nucleotide polymorphisms (SNPs), certain CNVs are linked to a higher risk for diseases like cancer, hereditary genetic disorders, autoimmune diseases, and others.
Here at Bionano Genomics, we equip clinical research labs with NxClinical, the most comprehensive and up-to-date solution for cytogenetics and molecular genetics in one system for analyzing and interpreting all genomic variants from microarray and next-generation sequencing (NGS) data.
This guide briefly introduces shallow sequencing CNV analysis, how it works, and how labs are taking advantage of it today.
A Brief Introduction to NGS-Based Copy Number Analysis
NGS technology advancements have significantly increased our capacity to identify various genomic variations, including SNVs, CNVs, and other structural variations. Recently, NGS data for CNV analysis has attracted attention due to improved algorithms and new technologies enabling concurrent detection of CNVs and SNVs.
NGS copy number methods hold an advantage over microarray techniques for detecting smaller CNVs. While arrays are more efficient for large CNVs, they are less sensitive to copy number changes below 50 kb. NGS uncovers small or novel variants that arrays may miss by offering a detailed genome view.
NGS-based CNV analysis allows labs to pinpoint variant locations accurately. High-resolution sequencing analysis complements arrays’ high throughput, facilitating a comprehensive genome understanding.
Understanding NGS-Based CNV Calling and Analysis
Four primary methods detect CNVs with NGS data:
- Read-Pair (RP)
- Split-Read (SR)
- Read-Depth (RD)
- Assembly (AS)
Each method targets a specific CNV form or size range, leading to breakpoint accuracy trade-offs. None are perfect, with varying advantages and disadvantages. Many labs combine methods, such as read-depths with read-pairs or split-reads, for a comprehensive analysis.
As Dr. Fen Guo, Clinical Laboratory Director at ElmerPerkins Genomics notes, the utility of these methods often hinges on the quality of the NGS data available.
“There’s a general sense that some methods are better than others—for example, that the split-read method isn’t effective for accurate breakpoint identification because of the nature of this methodology, while the read-depths can detect small or large CNVs in all types of regions in the genome. But in addition to recognizing the inherent differences between these methods and what they’re capable of, so much depends on the quality of the data—the coverage depths, the read lengths, and the captured region.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
Here’s a very brief overview of each method:
Read-Pair compares insert size between read-pairs and a reference genome. It detects medium-sized (100kb-1Mb) insertions and deletions but is insensitive to smaller events (<100kb). It’s not suitable for low-complexity regions with segmental duplication.
Split-Read uses paired-end sequencing reads where only one pair maps reliably. This method identifies breakpoints at the base-pair level, but limited in detecting large-scale variants (1Mb or longer).
Read-Depth is based on the correlation between coverage depth and copy number. It detects CNV dosage and works better for large-sized CNVs. It can detect medium-sized (100kb-1Mb) insertions and deletions but struggles with small (<100kb) events.
Assembly can theoretically detect all genetic variations if reads are long and accurate. It’s designed to identify structural variation but is less used in CNV detection due to high computational demands.
Watch our free webinar—Copy Number Variant Detection by NGS: Coverage, Uniformity & Resolution—to see Dr. Guo introduce the main methods utilized for calling CNVs using NGS data and share clinical cases that illustrate how the coverage and uniformity of NGS data contribute to the resolution of CNV calling.
Calling CNVs from Low-pass Genome Sequencing Data
Low-pass genome sequencing or low-resolution genome sequencing has been proposed as a cost-effective alternative to detect clinically significant copy number variations.
Compared to traditional genome sequencing at above 30x coverage, low-pass genome sequencing only needs to achieve 1-10x coverage depending on its requirement for the resolution of CNV detection.
Low-pass genome sequencing enables labs to have different tiers of assays to achieve various clinical targets. For example, if you wanted to totally cover deletion and duplication events as well as loss of heterozygosity, perhaps only 5x coverage is sufficient. If you’re only looking for larger CNVs, such as aneuploidy, you may only need 1x or 2x coverage.
In addition, Low-pass CNV detection can be used to identify CNVs that are difficult to detect using other methods—for example, in identifying rare deletions in the genome.
Dr. Guo sees low-pass sequencing continuing to emerge as a primary alternative to microarrays given the often equal or better performance and non-biased analysis.
“I do see low-pass sequencing becoming a more popular alternative to microarrays. For labs looking to call CNVs that don’t have a microarray platform, but do have a sequencing platform, I would strongly suggest they consider low-pass sequencing. In our experience, the performance and resolution are equal to arrays—and sometimes even better. With arrays, your sensitivity is limited to where your probes are located. Low-pass sequencing is basically the same platform as genome sequencing. Rather than 40x coverage, you’re running 5x or 8x coverage—trying to catch the larger CNVs. It’s also important to note that shallow sequencing is PCR-free which means it is non-biased sequencing. It’s uniform data laid out across the entire genome, which isn’t limited to certain regions like with microarray. You may be able to detect events you otherwise would have missed because of probe limitation.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
Watch our free webinar—Genome sequencing reveals cause of multi-generational split hand/split foot with long bone deficiency—to see how Dr. Raymond C. Caylor, Assistant Director, Molecular Diagnostic Laboratory at Greenwood Genetic Center, utilized genome sequencing and Bionano’s NxClinical software, to provide a diagnosis for a multi-generational family with split hand/split foot with long bone deficiency.
CNV Detection and Analysis Software for Shallow Sequencing
CNV detection from NGS data is challenging, and most standard NGS analysis software struggles to detect or visualize CNVs. These tools are often limited to specific variant types or focused on SNVs. This leaves labs with an incomplete genomic picture, hindering comprehensive investigations and results.
This software falls into two broad categories: homegrown tools and commercial software. Homegrown tools are custom-built systems, often integrated with free CNV tools. Commercial software offers ready-to-use solutions with CNV-calling capabilities.
Despite potential cost advantages, homegrown CNV tools have drawbacks, including:
- Narrow applicability: Focused on specific NGS data types, homegrown systems don’t easily integrate with multiple data types, leading to workflow inefficiencies and lost time.
- Expertise requirements: Developing a CNV tool requires bioinformatics specialists for database establishment, optimization, and maintenance. Most labs lack the resources for continuous refinement and development.
- Rapid obsolescence: With constant advancements in NGS, homegrown tools quickly become outdated without a dedicated development team.
Conversely, commercial CNV software allows teams to access efficient solutions without needing in-house bioinformatics or development expertise. These user-friendly tools often stay up-to-date with NGS advancements.
However, performance, capability, and ease of use can vary among CNV software, with some treating CNV analysis as an auxiliary feature, as Dr. Guo points out. As Dr. Guo explains, many of the commercial tools in use today treat CNV analysis as an add-on capability:
“From my experience using several software platforms, many commercial platforms that tout CNV analysis were built for SNV calling and interpretation. CNV calling was added on, but the primary interface is still designed for SNV analysis. Many labs needing to call CNVs need to interface with this data at the genomic level and get the whole picture—especially labs coming from the microarray world that want to use a familiar platform.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
Dr. Guo urges teams to be thoughtful when evaluating commercial tools against their particular needs—both today and tomorrow:
“You have to be very careful when thinking about the best commercial tool for the type of CNV calling you need to do. Think about the primary purpose you’ll be using it for. Are you only going to be using panels? Only exome data? Or do you think you’ll want software that analyzes all types of NGS data and connects the dots between them? Here at PerkinElmer, we use panels, exome, and genome data, which is why we use software [Bionano’s NxClinical] that covers everything.
Secondly, most CNV software will give you deletions, duplications, and copy numbers. But not all of them call AOH, which is important for imprinting disorders and cancer.
Thirdly, you have to consider the differences in analytical performance between software. You don’t want a high false-positive rate or false-negative rate.
And lastly—and most importantly for me—if you or anyone on your team is a naturally visual person, you need to look at the data visualization and user interface. It needs to be user-friendly and not get in its own way. Events should be easy to identify.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
In summary, when assessing commercial CNV calling software, remember:
- Commercial software generally outperforms freeware and homegrown tools across several dimensions.
- Compare each tool’s capabilities against your needs.
- Some commercial software may not support multiple NGS data types.
- Not all tools call both CNVs and AOH.
- False-positive and false-negative rates differ between tools.
- User interfaces can vary.
NxClinical for CNV Detection and Analysis from NGS Data
NxClinical is a comprehensive and up-to-date solution for cytogenetics and molecular genetics, enabling labs to analyze and interpret all genomic variants, including CNVs, from microarray and NGS data.
As a single-source software solution, NxClinical is platform-agnostic and accepts various data types, allowing clinical research laboratories to process CNVs, SNVs, AOH/LOH, and soon structural variants from one place. This comprehensive view of a sample’s genome significantly increases lab efficiency and confidence.
NxClinical delivers unparalleled CNV clarity and resolution to an otherwise challenging data type. It features two perfected algorithms for detecting CNV and AOH from nearly all NGS assays with high sensitivity and low false-positive rates. These algorithms are available within NxClinical, allowing labs to detect CNVs and AOH regions and visualize SNVs in context across all microarray and NGS platforms simultaneously, all from a single screen.
- The “Self-reference” algorithm is applicable to all WGS data regardless of sequencing depth.
- The “Multi-Scale Reference” (MSR) algorithm is also applicable to all NGS data. The MSR algorithm creates “virtual” bins with sizes proportional to the expected number of reads, offering high-resolution detection of events in areas of interest (e.g., exons) while also providing a useful genome-wide backbone.
Calling CNVs from Shallow Sequencing Data with NxClinical
With higher depth NGS, smaller CNVs can be detected and integrated with sequence variants to provide a holistic view of the sample.
The MSR algorithm can be applied to detect CNVs from shallow sequencing, including very low-level mosaic events seen in NIPS or ctDNA samples. The image below shows a sample with trisomy 21 detected using 1x WGS.
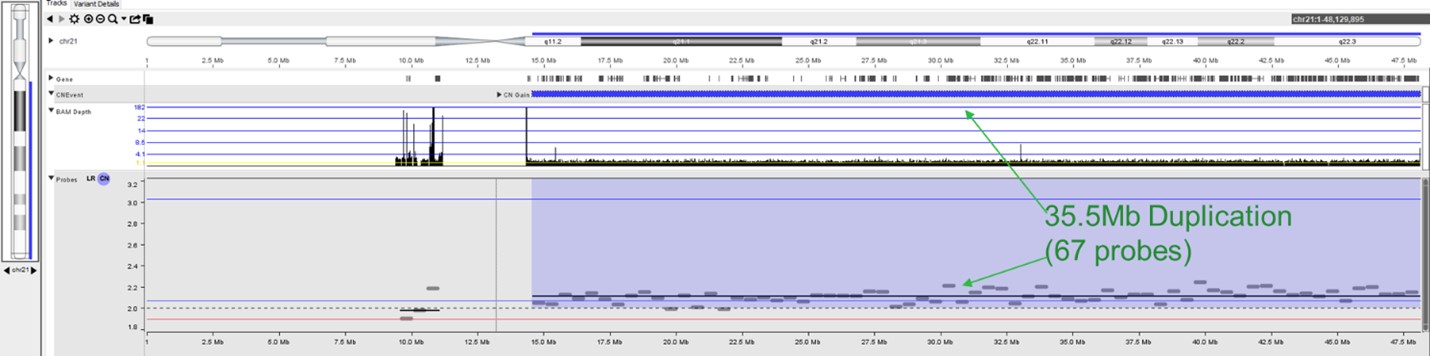
CNVs are an important contributor to disease and are required for accurate diagnosis. For clinical sequencing to be fully accepted as a replacement for microarrays and other widely used techniques, it must provide high-quality CNV information. NxClinical can easily and accurately provide that information from various approaches using NGS data.
Free tutorial for NxClincial users
Copy number analysis by NGS: Urban legend or true reality?
Are you an active NxClinical user considering an update? In this 25-minute webinar, Soheil Shams, Founder & CEO of BioDiscovery, a Bionano Genomics company, uses multiple example oncology cases to demonstrate the most effective workflow and case review benefits of the Knowledgebase in NxClinical 6.0.
Get the software trusted by renowned academic and commercial clinical labs to stay on top of demanding, time-sensitive workloads.
Book a free personalized demo to assess fit and see NxClinical in action. Let us know you’re interested and we’ll connect on an initial consultation to answer questions and dive a little deeper before demonstrating NxClinical—either with example data or your own.




