Blog
Identifying Statistically Significant Frequent Copy Number Alterations: STAC and GISTIC
When evaluating recurring copy number alterations in a larger data set, two questions come to mind: (1) What are the most frequent alterations and (2) are they statistically significant? The discovery edition of Nexus copy number software has two different tools to help answer this question, STAC and GISTIC. While both tools identify frequent, statistically significant copy number alterations, each does so through a unique method.
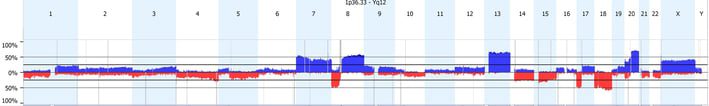
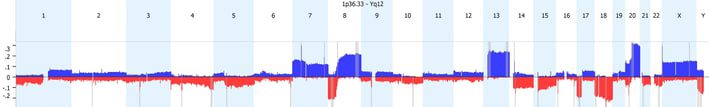
Significance Testing for Aberrant Copy number (STAC) (Sharon J. Diskin et. al. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006 Sep;16(9):1149-58. Epub 2006 Aug 9) is a method for testing the significance of DNA copy number aberrations across multiple array- experiments. In other words, the algorithm identifies sets of aberrations that are stacked on top of each other that would not occur randomly. To find these events, the aberrations in each arm of each chromosome are permuted and evaluated for the likelihood of an event to occur at any location at a particular frequency, as defined by the aggregate % cut-off option. Then a p-value cut-off is applied and significant regions are identified. This tool is located in the Results tab under the Aggregate section; click on Significant Peaks to apply. All significant regions will be shown on the Results – Genome view, while only those that pass the frequency threshold will be shown on the aggregate table.
STAC makes all of the calculations downstream of copy number and allele event calling. If the data has been processed via pairwise or matched-paired analysis, only the somatic calls will be included in this evaluation; background germline events, which were excluded during initial processing, will remain removed from this frequency significance testing. Also, any manual modifications to the calls (adding, removing or combining regions), will be included in this analysis. Any gender correction which has been applied to these samples will be taken into account.
The Genomic Identification of Significant Targets In Cancer (GISTIC) algorithm (Rameen Beroukhim et. al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci U S A. 2007 Dec 11;104(50):20007-12) is used to identify regions with a statistically high frequency of copy number aberrations over background aberrations. This tool was designed with cancer data sets in mind. Similar to STAC, GISTIC evaluates both frequency and significance to identify regions of interest. The G score measures both frequency of occurrence of the aberration and the magnitude of the copy number change (log ratio intensity) in each of the samples in the data set. Each location is scored separately for gains and losses. Then locations in each sample are permuted simulating data with random aberrations. This random distribution is compared to the observed statistic to identify scores that are significant. FDR multiple testing correction is applied to calculate a Q-bound significance score. Within each statistically significant region, a peak region is identified so that the region with a maximal G-score and minimal q-value is most likely to contain affected genes. This tool is located on the data set tab under Tools. Only regions which pass both the G-Score and Q-bound threshold cut-offs will be shown.
GISTIC is applied upstream of Nexus processing on the raw uploaded data. As a result, any modification to the raw data, including gender correction and manual modifications to calling, will not be included in this analysis. If the data has been processed by matched-paired analysis, the GISTIC output will be for somatic calls only. However, if any comparisons between tumor and normal were done downstream of the initial data upload, both germline and somatic calls will be included in this evaluation. Because GISTIC considers more data when assessing frequency and significance, specifically the magnitude of the log ratio intensity, it may be more sensitive at identifying potentially significant alterations that are present at a lower frequency.
If you have cancer samples and want to identify statistically significant regions of interest, which tool should you use? While GISTIC results on sex chromosomes may be misleading, STAC may overlook lower frequency significant regions of interest. Given the inherent strengths and weaknesses of each tool, a combined approach is most prudent.
Download our white paper, “Analyzing NGS Data For Copy Number Events,” to learn:
- How to use the BAM algorithm to obtain both copy number and allelic events from NGS data (unique and exclusive to Nexus Copy Number)
- Why this Read-Depth method is the most reliable and efficient means to extracting copy number information out of NGS data
- How the algorithm compares to other CNV estimating algorithms such as CoNIFER and XHMM

