Blog
Gene Panel CNV Analysis: A Brief Guide
Copy number variations (CNVs) refer to genomic modifications that lead to irregular copies of one or multiple genes. These alterations can be caused by structural genomic events, including duplications, deletions, translocations, and inversions.
Similar to single-nucleotide polymorphisms (SNPs), specific CNVs have been associated with increased vulnerability to diseases such as cancer, hereditary genetic disorders, autoimmune diseases, and more.
At Bionano Genomics, we provide clinical research laboratories with NxClinical, the most comprehensive and current solution for both cytogenetics and molecular genetics. This system enables the analysis and interpretation of all genomic variants from microarray and next-generation sequencing (NGS) data.
This guide briefly introduces gene panel CNV analysis, how it works, and how labs are taking advantage of it today.
A Concise Overview of NGS-Driven Copy Number Analysis
The advancement of NGS technology has significantly enhanced our capacity to identify various genomic variations, ranging from single nucleotide variants (SNVs) to CNVs and other structural variations. In recent years, the utilization of NGS data for CNV analysis has gained considerable interest thanks to the emergence of novel technologies and improved algorithms that allow for the concurrent detection of CNVs and SNVs.
NGS copy number methods possess a distinct advantage over microarray techniques in identifying smaller CNVs. Although arrays may be more effective in detecting larger CNVs, they generally have lower sensitivity for identifying copy number alterations smaller than 50 kb.
NGS uncovers small or new copy number variants that arrays frequently overlook by offering a detailed, base-by-base perspective of the genome.
NGS-based CNV analysis approaches also allow laboratories to pinpoint the exact location of a variant. The high-resolution examination facilitated by sequencing perfectly supplements the high throughput of arrays, enabling a thorough representation of the genome.
How NGS-Based CNV Calling and Analysis Works
There are four main methods of detecting CNVs with NGS data:
- Read-Pair (RP)
- Split-Read (SR)
- Read-Depth (RD)
- Assembly (AS)
Each of these four techniques excels at identifying a specific type or size range of CNV, which leads to a compromise in breakpoint precision. Each has its own pros and cons. To mitigate the limitations of each, many labs merge various techniques, such as combining read-depths with read-pairs or read-depths with split-reads, for a more comprehensive analysis.
As Dr. Fen Guo, Clinical Laboratory Director at ElmerPerkins Genomics notes, the utility of these methods often hinges on the quality of the NGS data available.
“There’s a general sense that some methods are better than others—for example, that the split-read method isn’t effective for accurate breakpoint identification because of the nature of this methodology, while the read-depths can detect small or large CNVs in all types of regions in the genome. But in addition to recognizing the inherent differences between these methods and what they’re capable of, so much depends on the quality of the data—the coverage depths, the read lengths, and the captured region.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
To give a little more background and tease out some of these important nuances, we briefly summarize each NGS CNV calling method below.
Read-Pair
The read-pair technique was the pioneer in showcasing the potential of NGS data for CNV detection. It operates by contrasting the insert size between actual sequence read-pairs with the anticipated size based on a reference genome. Labs utilizing this method can pinpoint CNVs by examining the discrepancy between mapped paired reads whose distances substantially deviate from the pre-established average insert size.
- The read-pair method can detect medium-sized (100kb to 1Mb) insertions and deletions from mapped data. However, it lacks sensitivity to small insertion or deletion events (<100 kb, or even intragenic deletions and duplications).
- This method is unsuitable for detecting CNVs in low-complexity regions with segmental duplication.
Split-Read
The split-read approach employs reads from paired-end sequencing, where only one pair has dependable mapping, and the other either wholly or partially fails to map to the genome.
The unmapped reads are a potential source of breakpoints at the single base-pair level. However, this method has limited ability in identifying large-scale sequence variants (1Mb or longer).
Read-Depth
The read-depth technique is founded on the assumption that a connection exists between a genomic region’s coverage depth and the region’s copy number.
- This method can detect the dosage of CNVs and works better on large-sized CNVs. This method can also detect medium-sized (~100kb to 1Mb) insertions and deletions from mapped data.
- However, this technique is not reliable for detecting small (<100kb) insertion or deletion events, and its sensitivity can vary depending on the platform and assay.
Assembly
In principle, all forms of genetic variation, including CNVs, can be identified through the assembly of short reads if the reads are long and accurate enough.
- This approach was developed to enhance structural variation identification. However, it is less frequently employed in CNV detection due to the intense strain it can place on computational resources.
Watch our free webinar—Copy Number Variant Detection by NGS: Coverage, Uniformity & Resolution—to see Dr. Guo introduce the main methods utilized for calling CNVs using NGS data and share clinical cases that illustrate how the coverage and uniformity of NGS data contribute to the resolution of CNV calling.
Calling CNVs from Gene Panels
A gene panel is a targeted set of genes sequenced in a patient to identify mutations that can cause rare diseases. Researchers typically choose the targeted set of genes based on their biological function and/or based on rare disease phenotypes. Several methods, including “capture” and “amplicon” sequencing, are used to sequence a panel of genes in patients with rare diseases.
Mutations identified by gene panels can include SNVs and CNVs. Thanks to recent technological advancements, the clinical research utility of gene panels has expanded greatly in the past decade and is now becoming more affordable for many clinical diagnostic laboratories worldwide.
- As Dr. Guo describes, compared to MLPA, which has long been considered the gold standard for CNV calling, gene panels offer a high-throughput alternative that enables more granular analysis.
- Gene panels offer higher coverage compared to MPLA, which means the sensitivity of CNV calls can be comparatively higher.
“The utility of Multiplex ligation-dependent probe amplification (MLPA) is limited due to the number of probes included in the kit. It is designed to multiplex up to approximately 50 probes, hence most suitable for one or a few smaller genes.
Gene panels enable you to detect the CNVs for all of the genes included in a given panel. Due to being deep-sequenced, the panel data often have high coverage depth, which increases the accuracy of CNV detection via the RD approach, although the fact that intronic regions are not included in the analysis may give a somewhat lower sensitivity to certain CNVs compared to using whole genome data.”— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
When calling CNVs from gene panels, Dr. Guo stresses that the specific performance and resolution one can achieve depends entirely on the data at hand—how that gene panel was designed.
“At PerkinElmer Genomics, we have a comprehensive DMD gene panel. When we designed this panel, we not only covered the exon regions but the entire DMD gene. This allows us to call out single-exon-level deletions and duplications. And we can identify breakpoints down to the nucleotide level.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
Dr. Guo’s central point for other labs: During the validation of a gene panel, balancing the sensitivity and specificity to provide better performance or resolution for CNV calling is critical.
Watch our free webinar—Genome sequencing reveals cause of multi-generational split hand/split foot with long bone deficiency—to see how Dr. Raymond C. Caylor, Assistant Director, Molecular Diagnostic Laboratory at Greenwood Genetic Center, utilized genome sequencing and Bionano’s NxxClinical software, to provide a diagnosis for a multi-generational family with split hand/split foot with long bone deficiency.
Software for Detecting and Analyzing CNVs from Gene Panels
Accurately detecting CNVs from NGS data has been a persistent challenge for clinical research laboratories. Most standard NGS analysis software tools struggle to efficiently identify or visualize CNVs. Their abilities are often restricted to certain variant types and sizes, or primarily focused on detecting SNVs.
In the absence of powerful and user-friendly CNV calling capabilities, labs are left with an incomplete view of genomic abnormalities, preventing them from thoroughly examining patient samples and providing comprehensive results.
Today’s software tools for detecting, analyzing, and interpreting CNVs from NGS data can be broadly divided into two categories: homegrown tools and commercial software.
- Homegrown tools are custom-made systems developed from the ground up and integrated with freely available online CNV tools.
- Commercial software are purpose-built systems labs purchase and integrate into their workflow with CNV-calling capabilities.
While homegrown CNV tools may offer cost benefits if a lab has highly specific and constant CNV calling requirements, they also present several drawbacks that can result in significant practical and efficiency expenses for a lab.
For example, homegrown systems and CNV freeware usually cater to a specific NGS data type exclusively. Handling multiple NGS data types—such as panels, whole-exome, and whole-genome data—requires using various tools that might not integrate smoothly or at all, leading to workflow inefficiencies and time costs for labs and patients.
Creating a homegrown CNV analysis tool also demands bioinformatics expertise. A team of bioinformatics specialists is needed to develop, optimize, scrape, and train a database for a robust CNV calling tool. The development effort can be immense before the system is ready for use, and most labs lack an in-house bioinformatics team for continuous maintenance and refinement.
Thirdly, homegrown CNV tools often become outdated rapidly. As NGS is a constantly evolving field, labs regularly have opportunities to enhance their genomic analysis speed and quality. However, without a development team to update their tools, labs may invest heavily in custom tools that quickly become obsolete.
On the other hand, commercial CNV software allows teams to benefit from efficiencies and features without needing in-house bioinformatics or development expertise. These tools are generally more user-friendly and stay up-to-date with the latest advancements in NGS capabilities. However, the performance, capability, and ease of use of CNV software can vary.
As Dr. Guo explains, many of the commercial tools in use today treat CNV analysis as an add-on capability:
“From my experience using several software platforms, many commercial platforms that tout CNV analysis were built for SNV calling and interpretation. CNV calling was added on, but the primary interface is still designed for SNV analysis. Many labs needing to call CNVs need to interface with this data at the genomic level and get the whole picture—especially labs coming from the microarray world that want to use a familiar platform.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
Dr. Guo urges teams to be thoughtful when evaluating commercial tools against their particular needs—both today and tomorrow:
“You have to be very careful when thinking about the best commercial tool for the type of CNV calling you need to do. Think about the primary purpose you’ll be using it for. Are you only going to be using panels? Only exome data? Or do you think you’ll want software that analyzes all types of NGS data and connects the dots between them? Here at PerkinElmer, we use panels, exome, and genome data, which is why we use software [Bionano’s NxClinical] that covers everything.
Secondly, most CNV software will give you deletions, duplications, and copy numbers. But not all of them call AOH, which is important for imprinting disorders and cancer.
Thirdly, you have to consider the differences in analytical performance between software. You don’t want a high false-positive rate or false-negative rate.
And lastly—and most importantly for me—if you or anyone on your team is a naturally visual person, you need to look at the data visualization and user interface. It needs to be user-friendly and not get in its own way. Events should be easy to identify.”
— Dr. Fen Guo, Ph.D., FACMG, FCCMG, Clinical Laboratory Director at PerkinsGenomics
NxClinical for CNV Detection and Analysis from NGS Data
At Bionano Genomics, we provide labs with a unified software solution to address these challenges.
NxClinical is the most all-encompassing and current solution for both cytogenetics and molecular genetics, enabling the analysis and interpretation of all genomic variants, including CNVs, from microarray and NGS data within one system.
- NxClinical is platform-agnostic. It accepts various data types that enable clinical research laboratories to process CNVs, SNVs, AOH/LOH (and soon structural variants)—all from a single place.
- These aberrations visualized in one software provide a complete picture of a sample’s genome, enabling labs to work significantly more efficiently and confidently.
- In short, NxClinical brings genuine CNV clarity and resolution to an otherwise difficult data type.
We’ve perfected two algorithms for the detection of CNV and AOH from almost all NGS assays with high sensitivity and low false-positive rates.
Both are available with NxClinical, the genomics software solution that enables labs to detect CNVs and AOH regions, and visualize SNVs in context, across all microarray and NGS platforms simultaneously—all from a single screen.
- One algorithm, the “Self-reference” algorithm, can be used for all WGS data regardless of sequencing depth.
- The second algorithm is the “Multi-Scale Reference” (MSR) algorithm that is also applicable to all NGS data. The MSR algorithm is able to create “virtual” bins with sizes proportional to the expected number of reads offering high-resolution detection of events in areas of interest (e.g. exons) while also providing a helpful genome-wide backbone.
Calling CNVs from Gene Panels with NxClinical
With higher depth NGS, smaller CNVs can be detected and integrated with sequence variants to provide a holistic view of the sample.
The MSR algorithm can be applied to any gene panel from a single gene (e.g. DMD test) to large panels having thousands of genes. The image below is from the Illumina TruSightTM Oncology 500 (TSO500) panel showing a somatic cancer profile.
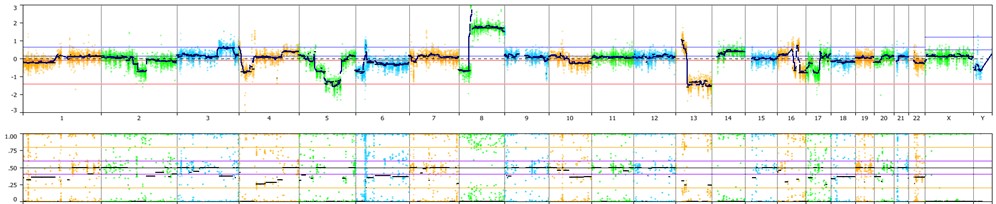
The cytogenetic complexity of the tumor sample is clearly evident with a large copy number gain of 8p and loss of a large section of 13q. Aberrations associated with genomic scarring, such as Loss of Heterozygosity (LOH), telomeric allelic imbalances (TAI), and large-scale state transitions (LST) can be visualized and manually called with confidence.
Free tutorial
Copy number analysis by NGS: Urban legend or true reality?
Are you an active NxClinical user considering an update? In this 25-minute webinar, Soheil Shams, Founder & CEO of BioDiscovery, a Bionano Genomics company, uses multiple example oncology cases to demonstrate the most effective workflow and case review benefits of the Knowledgebase in NxClinical 6.0.
Get the software trusted by renowned academic and commercial clinical labs to stay on top of demanding, time-sensitive workloads.
Book a free personalized demo to assess fit and see NxClinical in action. Let us know you’re interested and we’ll connect on an initial consultation to answer questions and dive a little deeper before demonstrating NxClinical—either with example data or your own.




