Blog
Gene Ontology Enrichment: Which Pathways or Processes are enriched in my data set?
During the course of your analysis with Nexus Copy Number, you may want to know about the specific pathways and processes that may be targeted by copy number alteration. We can think about this enrichment in two different respects: Whole genome (enrichment among selected samples across the whole genome) or specific regions (enrichment among selected regions of interest).
Whole genome enrichment identifies gene ontology (GO) terms enriched with copy number change over the whole genome, for all of the samples you have currently selected. While this type of analysis is typically recommended for samples with few changes overall, it can be run on any data set. When this analysis option is selected (found on the Results>Genome tab, click on Enrichments), enriched terms are identified and ranked; no additional settings are needed. This method is based on that described in the article Gene set enrichment analysis: A Knoelwdge-based approach for interpreting genome-wide expression profiles.
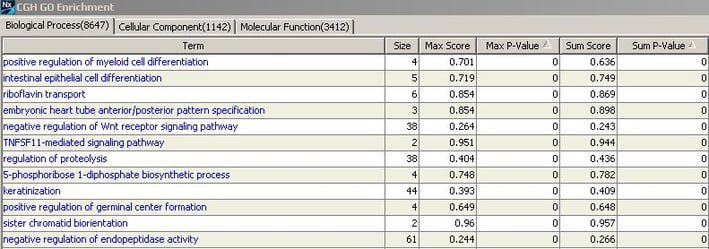
The results will display all GO terms along with high scores based on copy number gain OR loss (Max score) and on copy number gain AND loss (Sum score). Each score will have an associated p-value. By sorting based on significance, you can quickly identify any pathways or processes that may be enriched among all of the copy number changes in your data set. Note that you can further customize your return results by selecting additional enrichment data sets (File>Options>Enrichment Sets). And don’t forget, results displaying a “0” p-value are highly significant, past 3 decimal places. To find the actual significance in scientific notation, select Export and save your results.
What about finding significant pathway or process enrichment among comparison, concordance, GISTIC or STAC results (just to name a few)? Throughout your various downstream analyses, you will find the option to learn more about enrichment of these regions – anywhere you see the Enrichment button. Simply highlight some or all of the regions in blue (use the shift key to select a section or the control key to select specific results) and click on Enrichment. This is going to identify the GO terms enriched only within the regions you have selected.
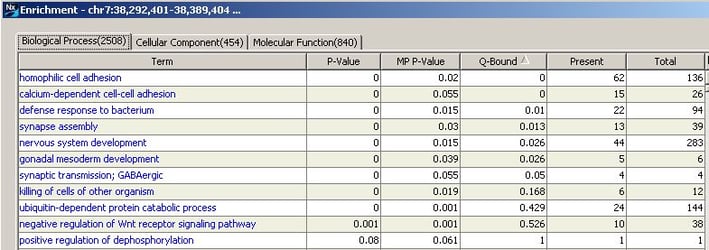
The results will display the significantly over-represented GO terms, the total number of genes in the term, and the number of genes present within the regions you defined. The p-value column will display the standard p-value or the probability of each particular gene being present in the set, treated independently of all other genes. The MP p-value displays the Markov process p-value or the probability of each gene being present in the set, taking into account genomic location. A copy number change in genes involved in the same pathway/process that are located nearby one another (and likely subjected to change by the same copy number event) would be weighted less heavily than copy number changes in genes involved in the pathway/process that are spread out throughout the genome. Q-bound significance for multiple testing correction by FDR results are also shown.
Want to know more about the genes involved in the pathway or process? Click on the blue hyperlink to get detailed information about the genes present in this GO term. Genes highlighted in yellow specify those present among the genomic regions you selected.
Need to generate some results for enrichment analysis? Check out our posts on Concordance and STAC and GISTIC.

