Blog
Comparison of CNV Detection from WES vs. SNP Microarray
Copy number variants have been implicated as drivers of many birth defects, developmental disorders, and even cancer.
The platform of choice to detect genome-wide CNVs has traditionally been microarray (including SNP arrays that can also detect copy-neutral LOH regions). However, since samples have often undergone sequencing to discover pathogenic sequence variants, labs want to exploit these data to also detect CNVs.
Here, we briefly compare the similarities and differences in CNV detection between WES and SNP microarray methods.
- Download our free white paper for a more in-depth comparison of CNV estimation results from various calling methods with data from WGS(<1x), WES (30x), cancer panels, and microarray.
- Watch our free webinar where Dr. Elan Hahn discusses how the team at Children’s Hospital of Los Angeles (CHLA) studied WES data and moved it one step further by implementing the use of BioDiscovery’s NxClinical software to analyze copy number variants (CNVs)—resolving ~9% of previously undiagnosed Mendelian conditions that had not been originally identified by WES, and in some cases chromosomal microarray.
A brief review of today’s CNV detection methods
Various methods for detecting CNVs from NGS data have come into maturity over the past few years, and are now routinely used in both research and clinical labs. Some of the more long-standing CNV detection methods include CoNVEX, CoNIFER, and XHMM.
But new methods have also emerged and become popular among labs, like the open-source CNVKit and our own BAM (multiscale reference or MSR) method, which offer comparable capabilities and are far more user-friendly than legacy methods.
Many older methods—like CoNIFER, xHMM, and QDNA—require strong bioinformatics expertise and use of the command line. And some are adept in only one arena (e.g. cancer or constitutional samples, data from WGS or targeted panels, etc.). Biodiscovery’s BAM MSR algorithm, by contrast, derives copy number and allelic event changes from WES, WGS, targeted panels, and low pass sequencing data, all from a convenient interface within our platforms.
- Download our free white paper for a comparison of CNV estimation results from BAM (multiscale reference) and other methods with data from WGS(<1x), WES (30x), cancer panels, and microarray.
WES vs. SNP microarrays
Perhaps the biggest substantive difference between these WES methods and SNP microarrays is the quality of the data post-processing. Here, microarrays, generally speaking, still have an advantage over WES for CNV detection specifically.
After the NGS data has been processed, WES methods essentially mimic a traditional microarray method. Each employs its own approach for creating so-called “pseudo probes” from the NGS reads. These reads are averaged in a certain bin or sliding window and divided by the number of reads in a reference sample (or group of reference samples if using our MSR method) to establish a log2 ratio value, which can then be used to estimate actual copy number.
SNP microarrays, by contrast, have actual probes. So, during an experiment or sample analysis, they receive an intensity value from those probes after labeling or staining, which, like with WES methods, is also compared to a reference probe. The idea behind the methods is quite similar.
Here at BioDiscovery, the BAM MSR method for CNV detection from NGS and microarray has been highly refined and is conveniently deployed through our research and clinical platforms, Nexus Copy Number and NxClinical.
A head-to-head comparison with five constitutional germline samples
In 2015, when the question of CNV detection from WES NGS data versus SNP microarrays was first being investigated, we ran a formal comparison to understand which technique yielded better results under certain conditions.
Using a data set of five constitutional germline samples that had been subjected to both WES and to a genome-wide SNP microarray, we compared the ability to detect CNVs between these platforms.
How the comparison worked
In this analysis, BAM files from whole-exome sequencing and Affymetrix SNP 6.0 SNP array results were downloaded for five germline TCGA colon adenocarcinoma samples.
- BAM files were processed for copy number variation estimation using CoNVEX and then uploaded into Nexus Copy Number v7.5 for visualization and downstream analysis.
- Affymetrix SNP 6.0 CEL files were uploaded directly into the software. Quadratic systematic correction was applied to all CEL files to correct for probe-localized GC content.
- Using the SNP-FASST2 algorithm, a significance threshold of 1E-8 was used; one-copy gain was called if the median probe log-ratio within a segment was greater than 0.18, a high-copy gain was greater than 0.6, a one copy loss was less than -0.18, and homozygous deletion was lower than -1.0.
When comparing the overlap of regions of change from copy number estimation with SNP array and WES, concordance of copy number estimation between methods is dependent on both quality and coverage between methods.
What we found
As shown below, two samples (3667 and 3672) had reduced quality from WES, and resulted in a much higher number of copy number calls, as compared to the other three sample pairs. Overall, SNP arrays produced far fewer copy number calls as compared to WES.
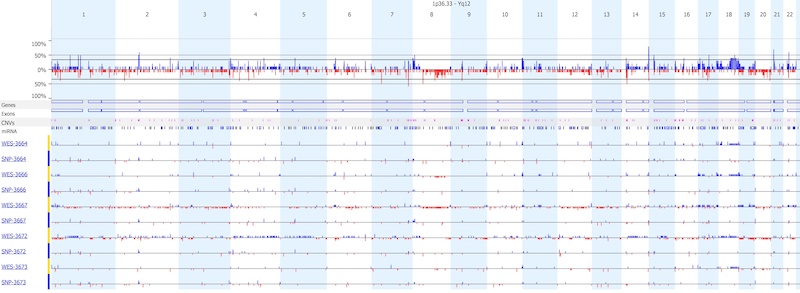
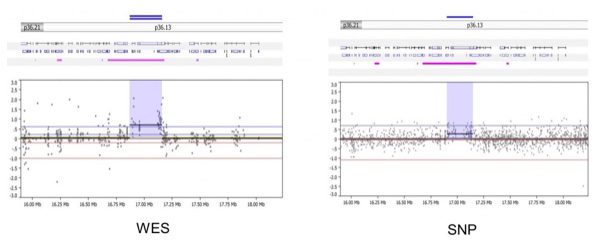
- WES and SNP arrays can detect concordant gene-level alterations, especially those that are longer and well covered by multiple exons (for WES) or probes (for SNP). Below, the WES (left) and SNP array (right) easily detect gain of a gene on chromosome 1 in matched sample 3664 on the top panel.
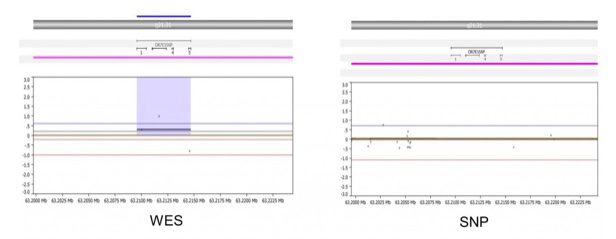
- In areas of poor SNP probe coverage, WES can detect events missed by the SNP array. As shown below, the WES (left) detects gain of OR7E156P on chr13, in sample 3664. SNP array (right) does not detect this change due to a lack of probe coverage on this gene.
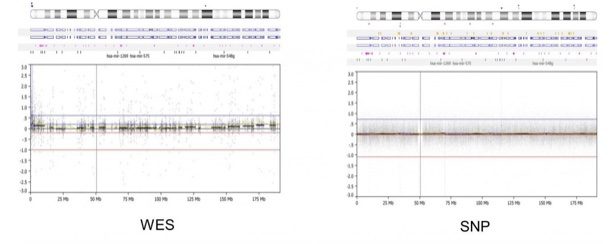
- However, the SNP array offers better overall genomic coverage and can detect intronic and intergenic alterations. The WES (left) is limited to genic/exonic coverage while the SNP array (right) offers more uniform coverage across the genome, in this example below of a whole-genome view of chromosome 4. Because of WES coverage, newer microarray designs have focused on exon regions by placing more probes. This way, arrays can essentially work backward to mimic WES and determine a so-called “exon-level” CNV call.
A few key takeaways (updated for 2022)
When we ran and first wrote about this comparison back in 2015, labs were just starting to adopt WES more and use microarrays less.
Back then, WES was run primarily to see sequence variants—mutations for one or a few bases. But labs simply didn’t have a way to call a copy number. At the time, this comparison was intended to explore some of the methods that had been proposed for detecting CNVs from WES data.
Since then, however, the trend away from a reliance on microarrays and toward greater adoption of NGS/WES has only accelerated. Now, many, especially newer labs don’t use microarrays at all, but still need to call copy numbers from their NGS data, making this discussion even more salient.
As BioDiscovery’s Dr. Zhiwei Che explains, some labs have radically refined their methods for getting copy numbers from WES to the point where they’ve discarded microarrays entirely.
“Since WES has grown in popularity, many labs have actually become so mature that they get many or all of the copy number results they need out of WES. So, they don’t use microarrays anymore.
Over the past few years, labs have done their own comparisons like the one we wrote about here to look at CNVs from WES versus CNVs from microarrays—and they’ve seen both methods yield a lot of the same calls.
And from exon regions specifically, many have seen even better calls from WES. We’re continuing to see labs replace their microarrays with WES for this very reason—and it’s why we developed and continue to refine our algorithm to call those copy numbers from NGS data.”
— Dr. Zhiwei Che, BioDiscovery
While microarrays remain the “gold standard” for CNV detection, the overwhelming adoption of WES in clinical labs has relegated the use of microarrays mostly as a way to validate or confirm the results of WES analysis. For labs continuing to use microarrays, those methods still offer a relatively easy means of calling CNVs.
However, for newer labs that don’t use microarrays, WES, when paired with a properly powerful analysis algorithm, provides the calling capabilities they need. In cases where confirmation is necessary, real-time PCR and FISH can be used to validate whether copy numbers are gained or lost in important genes.
Even for very small copy number changes, or changes that will only impact coding regions of the genome, WES has since proven itself just as reliable—and in some cases even more so—than microarrays.
“It’s fascinating and encouraging to see the results labs are seeing from running their own comparisons between WES and microarrays. Recently, a very large clinical lab ran one thousand samples through WES and microarrays side-by-side.
The results made them confident enough to retire their microarrays once they saw that with the right platform powered by the right algorithms, they can detect both sequence variants and CNVs from their WES data—rather than only getting CNVs from their arrays. Anecdotes like this speak to why WES is so popular.”
— Dr. Zhiwei Che, BioDiscovery
Dr. Che also notes that while it was once best practice to use multiple algorithms to estimate copy number variation from samples due to variance between algorithms, many methods (such as MSR and CNVKit) achieve a level of reliability that makes it unnecessary to run additional algorithms to confirm results.
“Unlike years ago, most labs don’t have to run so many different algorithms to confirm their CNV estimations.
Many labs using Biodisocvery’s platforms—NxClinical or Nexus Copy Number—only use our MSR to get their results; they don’t use other methods to confirm it because it’s been validated clinically from microarray and MCR methods.”
— Dr. Zhiwei Che, BioDiscovery
For labs using fewer or just a single CNV calling algorithm, it’s still important to consider batch effects when evaluating larger data sets.
“Microarray manufacturers offer reference files labs can use for baselines for establishing log ratios to get copy numbers. But with NGS, there’s no such universal reference file. So, different labs may run the same sample and get slightly different results depending on the technique and the reagent lots. Because many labs have the same workflow and personnel, we usually recommend labs run their normal samples through their workflow, and then build their own references.”
— Dr. Zhiwei Che, BioDiscovery
Copy number estimation from WES results can be applied to a number of different pathogenic diseases, such as postnatal genetic diseases, rare diseases, and oncology.
For germline diseases, including autism, developmental and neurological disorders—and for somatic cancers without a matched normal comparison, labs should consider using our MSR method for copy number estimation.
For somatic cancers with a matched normal comparison, ngCGH would likely still be the optimal method. NxClinical and Nexus Copy Number support visualization and downstream analysis from all of these copy number estimation algorithms.
Detect and display CNVs—all from one place
Detect CNVs and AOH regions, and visualize SNVs in context across all microarray and NGS platforms simultaneously—all from a single screen.
BioDiscovery’s MSR algorithm powers case review via NxClinical and research via Nexus Copy Number for detecting CNVs from NGS and/or microarrays—and displaying them along with SNVs for maximum context.
- NxClinical is the most comprehensive single-software cytogenetics and molecular genetics solution for analyzing and interpreting CNV, SNV, and AOH data across all platforms for patient samples. Labs using it simplify their case review workflows and make the right calls in record time. Learn more about NxClincial »
- Nexus Copy Number, also powered with the gold-standard CNV calling algorithm, derives copy number and BAF from a variety of NGS data (WES, WGS, targeted panel, and shallow sequencing) and microarray data. Nexus Copy Number is a multifaceted desktop software for rapid discovery of genomic alterations. This platform-agnostic software accepts data from various manufacturers and technologies including the latest platforms: Infinium GSA and CytoScan XON. Learn more about Nexus Copy Number »
Check out this webinar for more background on each of the algorithms mentioned above.
Get the software trusted by renowned academic and commercial clinical labs to stay on top of demanding, time-sensitive workloads
Book a free personalized demo to assess fit and see NxClinical or Nexus Copy Number in action.
Request a free demo and we’ll connect on an initial consultation to answer questions and dive a little deeper before demonstrating NxClinical or Nexus Copy Number—either with example data or your own.
*This software is for research use only. It is designed to assist clinicians and it is not intended as a primary diagnostic tool. It is each lab’s responsibility to use the software in accordance with internal policies as well as in compliance with applicable regulations.




